IMD
Configuration File Postprocessing
For the postprocessing of IMD configuration files, IMD contains a group of utility programs with a common usage model:
| imd_angle | compute angular distribution function |
| imd_cna | perform Common-Neighbour Analysis |
| imd_conn | compute connection matrix |
| imd_coord | compute coordination numbers |
| imd_elco | compute elastic moduli |
| imd_pair | compute pair distribution function |
| imd_ps | create PostScript picture |
| imd_ring | compute ring statistics |
| imd_strain | compute strain tensor from displacements |
| imd_stress | compute stress tensor from IMD stress output |
| imd_torsion | compute torsion angle distribution function |
These programs are compiled as follows:
gmake imd_utility[_option][_2d]where utility is the name of the program. Some programs support further options which can be appended to the compilation target with the component _option. By default, a program for 3d is compiled. Most programs support 2d versions which can be compiled using the additional component _2d in the compilation target.
The programs are run by giving the program name and one or more command line options. The following options are supported by all utility programs.
| Option | Values | Default | Description |
|---|---|---|---|
| -p | parameter_file | - | Specifies the IMD parameter file. This option is mandatory. |
| -r | integer >= 0 | - | Specifies the restart parameter. The corresponding checkpoint file is chosen as input file and also the iteration file is read in. If this parameter is not present, the file specified by the IMD paramter coordname is taken as input file. |
| -A | integer >= 0 | - | The corresponding avpos outfile is chosen as input file and also the avpos iteration file is read in. |
| -v | - | - | All virtual types are treated as being different. In this case, the number of types N is vtypes-1. If this option is not used, all atom types are taken modulo ntypes, the number of different real atom types, and N equals ntypes-1. |
Most of the programs produce output files. The name of these output files are the names of the input files with program specific suffixes. If checkpoint files are used as input files, the suffix .chkpt is omitted.
imd_angle
Computes angular distribution histograms of IMD configurations.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -n | integer > 0 | 1000 | Specifies the number of slots in the angular distribution histogram. |
| -e | real > 0.0 | 1.0 | Specifies the maximal radius for which angles between neighbouring atoms are computed. |
| -a | real >= 0.0 | 0.0 | Specifies the minimal radius for which angles between neighbouring atoms are computed. |
The output file name of imd_angle has the suffix .angle. For every slot in the histogram, one line is written:
phi g_000 g_001 ... g_00N g_011 ... g_01N ... g_0NN g_100 ... g_NNN
The first entry phi is the angle with range [0,180], and
the remaining entries are the histogram values for the different
combinations of atom types. The histogram entry g_ijk is
the normalized number of times the angle phi occurs
between the atoms of type i, j, and k
where the angle is centered at i. g_ijk is
normalized with respect to the total number of angles computed. In
the case of 2 types of particles, for example, the histogram has
the following format:
phi g_000 g_001 g_011 g_100 g_101 g_111
imd_cna
Performs the Common-Neighbour Analysis (CNA) for IMD configuration files.
The CNA classifies pairs of atoms according to their local environment. (J.D.Honeycutt and H.C.Andersen, J.Phys.Chem. 91 (1987) 4950, A.S.Clarke and H.Jónsson, Phys.Rev. E47 (1993) 3975)
A set of four indices ijkl (pair type) is used to characterize a pair of atoms having common nearest neighbour atoms where nearest neighbour atoms are defined by a cutoff distance. The indices have the following meaning:
| i | 1 if the atom pair consists of nearest neighbour atoms 2 otherwise |
| j | number of common nearest neighbour atoms |
| k | number of bonds between the common nearest neighbour atoms where bonded atoms are nearest neighbour atoms |
| l | number of bonds in the longest continuous chain formed by the k bonds between common nearest neighbours |
The cutoff distance can be given for all pairs of atom types by the parameter r_cut in the parameter file. Alternatively, a common cutoff distance for all types can be specified by the option -e.
imd_cna gives a list of the found pair types along with their occurrences to standard output in the following form:
pair type occurrence relative occurrence -------------------------------------------------------- ijkl n_ijkl t_ijkl ... ... ... ... ... ...where n_ijkl is the number of times the pair type ijkl occurs and t_ijkl is n_ijkl divided by the total number of pairs.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -a | real >= 0.0 | 0.0 | Specifies the minimal radius in the histogram of the radial distribution function. Only works in conjunction with the option -g. |
| -e | real > 0.0 | 1.0 | Specifies the cutoff distance for which atom pairs are considered as nearest neighbours. If the option -g is used, the maximal radius in the histogram of the radial distribution function is twice this value unless the option -f is set. |
| -f | - | - | Consider only pairs of atoms being nearest neighbour atoms, i.e., consider only pair types 1jkl |
| -g | - | - | Write the decomposition of the radial distribution function
(RDF) with respect to the pair types to a file with suffix
.rdf. The output format of the histogram is
r total_rdf t1 t2 ...
where r is the radius, total_rdf is the total
RDF, and t1, t2, ... are the contributions to the
RDF from the respective pair types (given in the header line of the
.rdf file). All RDFs are normalized with respect to the
number of pairs. |
| -l | real, real, (real) | -infty, -infty, (-infty) | Use a partial box with lower coordinates given by three (two) numbers. Only pairs of atoms where at least one atom has larger coordinates are considered. |
| -n | integer > 0 | 1000 | Specifies the number of slots in the histogram of the radial distribution function. Only works in conjunction with the option -g |
| -w | integer ijkl | - | Write all atoms belonging to pairs of type ijkl to a file with suffix .ijkl.cna. The output format is the standard format for IMD configurations. |
| -W | 4 integers i j k l |
- | Similar to option -w, but the pair type ijkl is given with spaces |
| -u | real, real, (real) | infty, infty, (infty) | Use a partial box with upper coordinates given by three (two) numbers. Only pairs of atoms where at least one atom has smaller coordinates are considered. |
imd_cna can be used to detect dislocations and stacking faults in crystalline structures. For example, fcc and hcp structures are characterized by the following pair types.
| fcc | hcp |
|---|---|
| 1421 2100 2211 2444 |
1421 1422 2211 2333 2444 |
imd_conn
Computes the connection matrix.
The following special option can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -n | integer > 0 | 10 | Specifies the maximal number of neigbours. |
imd_coord
Computes coordination numbers of IMD configurations. imd_coord needs the specification of cutoff radii for the atom types. If the component _tersoff is used in the compilation target, the cutoff radii for the Tersoff potential are taken from the parameter file and the coordination numbers are computed slightly differently (see below).
imd_coord writes two tables to standard output. The first table contains the relative occurrences c_ij of bonds between atoms of type i and j. It has the following format:
Bond Occurrence
00 c_00
01 c_01
...
0N c_0N
10 c_10
...
NN c_NN
The second table contains the occurrences occ_i in percent of the coordination number i up to the coordination number cmax. It has the format
Coordination Occurrence
0 occ_0
1 occ_1
...
cmax occ_cmax
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -e | real > 0 | 1.0 | Cutoff radius. Specifies the maximal distance for which neighbouring atoms are taken into account. |
| -a | real >= 0 | 0.0 | Specifies the minimal distance for which neighbouring atoms are taken into account. Does not work if the Tersoff potential used. |
| -C | integer > 0 | 10 | Specifies the maximal coordination number cmax the occurence of which is computed and written to standard output. |
| -g | - | - | Write the table of the occurrences of the coordination numbers into a file with suffix .gcoord. |
| -l | - | - | Write the local coordination numbers for each atom into a file
with suffix .coord. The output format is
n type x y (z) c_tot c_0 c_1 ... c_N
where n is the atom number, type is the type of
the atom, x,y,(z) are the coordinates of the atom,
c_tot is the total coordination number, and c_i
is the coordination number corresponding to neighbour atoms of type
i. |
| -E | - | - | Write the potential energy into the file .coord. This
option works only in conjunction with the option -l. The
output format is
n type x y (z) c_tot c_0 c_1 ... c_N E_pot
|
| -u | - | - | Set the cutoff function fc of the Tersoff potential equal to unity within the cutoff radius (see below). |
If imd_coord is compiled with the _tersoff component, the value of rmax is computed from the cutoff parameters of the Tersoff potential. Furthermore, the coordination numbers NiX are computed by default as
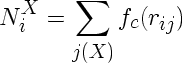
where the sum is over all neighbour atoms j of atom i with type X and fc is the cutoff function of the Tersoff potential.
imd_elco
Computes the stress tensor, the tensor of the elastic moduli, and the pressure derivative of the bulk modulus of IMD configurations using the interatomic potentials.
The following potentials are currently supported:
- Tabulated pair potentials. This is the default.
- EAM potentials. In this case, the component _eam has to be used in the compilation target.
- Many-body potentials. If the components _tersoff, _stiweb, or _keating are used in the compilation target, the Tersoff, the Stillinger-Weber, or the Keating potential are used, respectively. Many-body potentials only work in 3d.
By default, imd_elco computes the global stress tensor sigma_ij, the global tensor of the elastic moduli C_ab, and the global pressure derivative of the bulk modulus B'. These values are written to standard output along with the atom number N , the total volume V, the volume per atom v, the potential energy E, the total bulk modulus B and the pressure p.
imd_elco assumes that eV and Å are used as units of energy and length. The output data are written in these units and GPa for convenience. If other units are used, the units eV and Å should be ignored.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -s | - | - | Write the local stress tensor to a file with suffix
.stress. The output format is
number type x y (z) s_xx s_yy (s_zz s_yz s_zx) s_xy vol_voronoiwhere number is the atom number, type is the atom type, x,y(,z) are the coordinates of the atoms, s_ij are the components of the stress tensor, and vol_voronoi is the volume of the Voronoi cell. The stress tensor is normalized with the volume of the Voronoi cell of the corresponding atom. |
| -e | real > 0 | 1.0 | Specifies the maximal distance for which neighbouring atoms are taken into account in the computation of the Voronoi cells. It should be larger than the potential cutoff. This option is only needed in conjunction with the options -s, -m, or -M. |
| -m | - | - | Write the local elastic moduli to a file with suffix
.elco. The output format is
number type x y (z) B C_11 C_12 (C_44)The elastic moduli are normalized using the volumes of the Voronoi cells. |
| -M | - | - | Write the complete tensor of the local elastic moduli to a file
with suffix .elco. The output format is
number type x y z B C_11 C_12 C_13 C_22 C_23 C_33 C_44 C_45 \ C_46 C_55 C_56 C_66 C_14 C_15 C_16 C_24 C_25 C_26 C_34 C_35 C_36 |
| -w | integer ab | - | Write selective component C_ab (Voigt notation) of the tensor
of the local elastic moduli to a file with suffix
.c_ab.elco. The output format is
number type x y (z) C_ab |
imd_pair
Computes radial distribution histograms of IMD configurations.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -n | integer > 0 | 1000 | Specifies the number of slots in the pair distribution histogram. |
| -a | real >= 0.0 | 0.0 | Specifies the minimal radius in the histogram. |
| -e | real > 0.0 | 5.0 | Specifies the maximal radius in the histogram. |
r d_00 d_01 ... d_0N d_11 ... d_NN f_00 f_01 ... f_0N f_11 ... f_NN
The first entry r is the radius, the remaining entries are
the histogram values for the different combinations of atom types.
The histogram entry d_ij is the number of times the
distance r occurs between atoms of types i and
j, divided by the total number of atoms, and f_ij =
d_ij/(4*pi*r^2) is a normalized radial distribution.
imd_ps
Creates (vector) PostScript files from IMD configurations.
imd_ps produces ball and stick visualizations of IMD configuration files. For the drawing of the bonds, cutoff radii for all atom types have to be given in the parameter file by the parameters r_cut or ters_r_cut. Alternatively, the option -e can be used.
The geometrical setup of the projection (3d) is shown in the following figure.
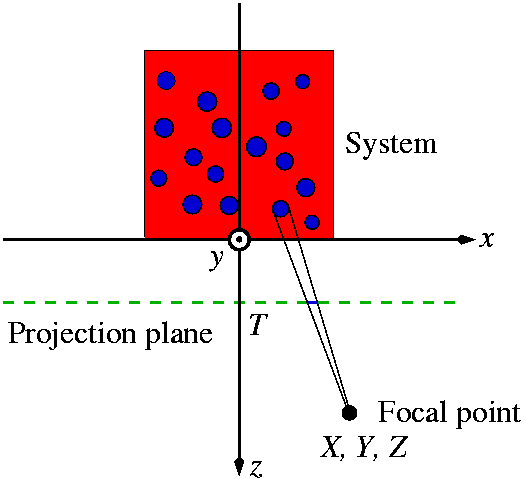
imd_ps allows many optional parameters affecting the visualization (Grey background means that the option is available only in the 3d version):
| Option | Values | Default | Description | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| -a | 0,1 | 0 | Draw coordinate axes. The axes are drawn into the bounding box of the unrotated system (option -u) or the simulation box (option -o). The x axis is red, the y axis is yellow, and the z axis (3D only) is green. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -b | real, [-1,1] | -0.3 | Brightness of bond colors. Positive values mean bright colors and negative values mean dark colors. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -B | real, >= 0.0 | estimated | Radius of cylinders representing bonds in real (IMD) units. The value of -B must be smaller than the value of -R. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -c | real, >= 0.0 | 20 | Boundary strip around bounding box (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -C | integer | 1 | Color of atoms. Negative numbers produce greyscales. The value
0 corresponds to white. imd_ps predefines 9 different colors
represented by integers as follows:
The option -C allows positive integer values with up to 9
figures which denote the colors of atom types 0,1,2,.... The
default colors correspond to the value 123456789.
|
||||||||||||||||||||||||||||||||||||||||||||||||
| -d | real, [0,1] | 0 | Brightness of atom and bond colors with depth. 1 corresponds to black atoms and bonds in the background, 0 means the same colors for all atoms and bonds. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -D | integer, >=0 | 0 | Represent types of atoms by different radii of the spheres. As
in the case of the atom colors given by the parameter -C,
the association of atom types with sphere radii is done by an
integer with up to 9 figures. The encoding is given by the
following table:
The second row gives the radii of the atoms in units of the largest radius specified by the parameter -R. In the case of the number 9 corresponding to the radius 0, no atoms of the corresponding type are drawn and the bonds of this type are also omitted. |
||||||||||||||||||||||||||||||||||||||||||||||||
| -e | real > 0.0 | 1.0 | Specifies the maximal distance for which neighbouring atoms are taken into account in the drawing of bonds. Only works, if no cutoff length is given in the parameter file. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -E | integer | - | Color encoding of the potential energy as specified in the 10th
column of an IMD configuration file. The value of -E
determines the colors used in the encoding. Each figure of
-E corresponds to a color as given in the following table:
The sequence of figures in E then corresponds to a path in the rgb cube which is used to color encode the potential energy from the lowest energy to the highest energy. If only one positive figure is given, a path with two colors is taken where the second color is white. Negative numbers correspond to preselected paths as follows:
The spectrum of the used color encoding can be plotted with the additional option -k. |
||||||||||||||||||||||||||||||||||||||||||||||||
| -f | integer | 9 | Draw bounding box with linewidth given by the option -F. A negative value means no boundary box is drawn. Positive values correspond to colors according to the color scheme of the option -C</>. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -F | real | 2.0 | Linewidth of bounding box (in pt). Negative values correspond to dashed lines. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -g | real, >=-1.0 | 0.0 | Background color. White by default. Positive integer values correspond to colors according to the color scheme of parameter -C. Figures after the comma mean a shading of the corrresponding color. Negative values from -1 to 0 correspond to a shading from black to white. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -G | integer, >0 | - | Draw color encoding of the 2nd data column (after positions and velocities) of input file. Similar to the option -E. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -h | real, >=0 | - | Produce eps files with the given height in cm. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -H | real, >=0.0 | 0.1 | Linewidth used for borders of atoms, bonds, and bond separators (in pt). See also the options -L, -l, and -P for separate adjustments. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -i, -j | real | 0, 0 | Translation in x and y direction. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -I | real, >0 | 1.0 | Brightness of shading of atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -J | real, [-1,1] | 0 | Brightness of atom colors. Positive values mean bright colors and negative values mean dark colors. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -k | real | - | Draw a spectrum of the colors belonging to the different color encodings. The spectrum is placed below the bounding box, separated by a line. The Linethickness is given by the value of the option -K (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -K | - | - | Write a file with all settings of the options. The filename is the output filename with the extension .eps.opt. The .eps.opt file contains the complete command to run imd_ps with the current options. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -l | real, >=0.0 | 0.1 | Linewidth used for bond borders (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -L | real, >=0 | 0.1 | Linewidth used for atom borders (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -m | real, >=0 | 1 | Zoom. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -M | real, >=0 | 1 | Parameter to enhance the shrinking of atoms and bonds with depth. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -n | real >=0 | - | Draw lightshadow. n determines the linewidth (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -N | real, [0,1] | 1 | Draw bonds as needles. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -o | integer | -1 | Draw simulation box with linewidth given by the option -O. A negative value means no simulation box is drawn. Positive values correspond to colors according to the color scheme of the option -C. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -O | real | 1.0 | Linewidth used for simulation box (in pt). Negative values correspond to dashed lines. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -P | real >=0 | 0.1 | Linewidth used for bond separator (in pt). | ||||||||||||||||||||||||||||||||||||||||||||||||
| -q | integer | -1 | Draw border to boxes with linewidth specified by the option -Q. A negative value means no border is drawn. Positive values correspond to colors according to the color scheme of parameter -C. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -Q | real | 1.0 | Linewidth used for border of boxes (in pt). Negative values correspond to dashed lines. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -R | real, >= 0.0 | estimated | Radius of spheres representing atoms in real (IMD) units. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -s | real, >=0 | 1 | Scaling of the picture. In contrast to the zoom -m, the size of the entire picture is also scaled. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -S | real, >=0 | 45 | Shading of atoms. The value of -S corresponds to the angle of the light position. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -t | integer | - | Draw color encoding of kinetic energy. Similar to the option -E. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -T | real, >= 0.0 | estimated | z-coordinate of projection plane (in real (IMD) units). The value of -T must be smaller than the value of -Z. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -u | integer | -1 | Draw bounding box of unrotated sytem with linewidth U. A negative value means no simulation box is drawn. Positive values correspond to colors according to the color scheme of the option -C. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -U | real | 1.0 | Linewidth used for bounding box of unrotated system (in pt). Negative values correspond to dashed lines. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -V | integer | 0 | Color of bonds. The value 0 means the bonds are colored like the atoms. Other values correspond to colors according to the color scheme of the option -C. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -w | real, >=0 | - | Produce eps files with the given width in cm. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -W | real, >=0 | - | Produce wireframes with the specified line width (in real (IMD) units). If the atom radius given by the option -R is larger than the value of -W, atoms are drawn as specified by the corresponding options. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -x, -y, -z | real, real, real | 0, 0, 0 | Rotation angles about x, y, and z axis (only z axis in 2D version) of IMD configuration in degrees. | ||||||||||||||||||||||||||||||||||||||||||||||||
| -X, -Y, -Z | real, real, >0 | 0, 0, estimated | Location of focal point of perspective projection (in real (IMD) units). | ||||||||||||||||||||||||||||||||||||||||||||||||
imd_ring
Computes ring statistics of IMD configurations.
imd_ring searches all shortest closed paths in a configuration and gives a statistics. It only works with open boundary conditions.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -e | real > 0.0 | 1.0 | Specifies the maximal distance for which neighbouring atoms are taken into account. If the component _tersoff is given in the compilation target, the cutoff radii of the Tersoff potential are employed. |
| -l | integer > 0 | 10 | Specifies the maximal length lmax of rings that are computed. |
| -n | integer > 0 | 6 | Specifies the maximal number of neighbours for which memory of neighbour tables is allocated. In the case of the Tersoff potential, this value is taken from the parameter file. |
imd_ring writes a table to standard output with the following format:
Ring length Total occurrence Relative occurrence
3 n_3 p_3
4 n_4 p_4
. . .
. . .
. . .
lmax n_lmax p_lmax
where n_i is the number of rings of length i and
p_i is n_i divided by the total number of rings
up to the length lmax. imd_ring writes the table of
the relative occurrences of the ring lengths into a file with
suffix .ring.
imd_strain
Computes the strain tensor from the atomic displacements.
The displacements must be given in a file with the following format:
x y (z) dx dy (dz)
where x, y, z are the positions of the
atoms and dx, dy, dz are the
displacements. The displacement file must have the name
<outfiles>.dsp. Alternatively, the displacement file
can be created using the IMD option disloc. Then the restart
parameter -r<nn> should be given where
<nn> is the number of steps corresponding to the
displacement file.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -e | real > 0.0 | 1.0 | Specifies the maximal distance rmax for which neighbouring atoms are taken into account in the computation of the derivatives of the displacements. |
| -c | real > 0.0 | rmax | Specifies the width of the cells in the cell decomposition used in the computation. For very large systems, it is advisable to use a value larger than rmax |
The output file has the following format:
n x y (z) e_xx e_yy (e_zz) (e_yz) (e_zx) e_xy
where n is a number (not the IMD atom number), x,
y, z are the coordinates, and e_ij are
the components of the strain tensor.
imd_strain only computes the strain tensor if an atom possesses more than two neighbours. Atoms with less than three neighbours are omitted from the outfile.
imd_stress
Computes the stress tensor from an IMD .press file.
IMD determines the (raw) stress tensor by evaluating the virial corresponding to the atomic forces. imd_stress computes the true stress tensor by dividing the raw stress tensor of each atom by the volume of the Voronoi cell of the respective atom. The raw stress tensor must be given in a file with the following format:
x y (z) s_xx s_yy (s_zz) (s_yz) (s_zx) s_xy
where x, y, z are the positions of the
atoms and s_ij are the components of the raw stress
tensor. The stress file name must have the suffix .press.
Alternatively, the raw stress file can be created using the IMD
option stress (with the parameter vector press_dim
set equal to zero). Then the restart parameter -r
<nn> should be given where <nn> is
the number of steps corresponding to the stress file.
The following special option can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -e | real > 0.0 | 1.0 | Specifies the maximal distance for which neighbouring atoms are taken into account in the computation of the Voronoi cells. |
The output file has the following format:
x y (z) s_xx s_yy (s_zz) (s_yz) (s_zx) s_xy v
where s_ij are the components of the true stress tensor of
the atom with coordinates x, y, z and
v is the volume of the corresponding Voronoi cell.
imd_stress only computes the stress tensor if the Euler relation for a Voronoi cell is fullfilled. Otherwise, atoms are removed from the outfile.
imd_torsion
Computes torsion distribution histograms of IMD configurations.
The following special options can be used:
| Option | Values | Default | Description |
|---|---|---|---|
| -n | integer > 0 | 1000 | Specifies the number of slots in the torsion distribution histogram. |
| -e | real > 0.0 | 1.0 | Specifies the maximal radius for which torsion angles between neighbouring atoms are computed. |
The output file name of imd_torsion has the suffix .torsion.
For every slot in the histogram, one line is written:
phi g_0000 g_0001 ... g_000N g_0011 ... g_001N ... g_00NN g_0100 ... g_NNNN
The first entry phi is the angle with range [0,180], the
remaining entries are the histogram values for the different
combinations of atom types. The histogram entry g_ijkl is
the normalized number of times the torsion angle phi
occurs between the atoms of type i, j,
k, and l.
g_ijkl is normalized with respect to the total number of torsion angles computed. In the case of 2 types of particles, for example, the histogram has the following format:
phi g_0000 g_0001 g_0011 g_0100 g_0101 g_0110 g_0111 g_1100 g_1101 g_1111