IMD
MEAM Potentials (option meam)
The Modified Embedded Atom Method was formulated by Baskes (M.I.Baskes, Phys.Rev.B 46 (1992) 2727, M.I.Baskes, Mater.Chem.Phys. 50 (1997) 152). It consists of a generalization of the EAM potentials by including angular terms.
The total energy for the MEAM potentials have a form similar to the EAM potentials:

Here Fi is the embedding energy function, rhoi is the electron density at the position of atom i, phiij is a pair potential, and Sij is the screening function defined below.
For MEAM potentials, the electron density is given by

where the parameters t(s)i are weight factors and the quantities rho(s)i, s=0,...,3, are partial electron densities defined by
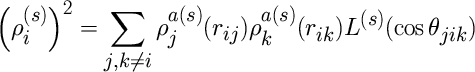
The functions L(s)(z) are Legendre polynomials, L(0)(z) = 1, L(1)(z) = z, L(2)(z) = z2-1/3, L(3)(z) = z3-3/5z. The atomic electron density functions from atom j at a distance rij from atom i are
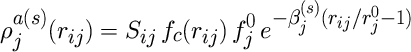
Here, f0i, r0i, and beta(s)i are parameters, fc is a cutoff function, and Sij is the screening function.
The many-body screening function Sij between atoms i and j is defined as the product of the screening factors Sikj due to all other neighbour atoms k of i and j,
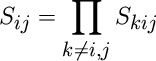
Skij is given by

where C1kij and C0kij are parameters and the function gc is defined as
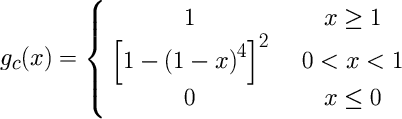
The cutoff function fc used in the atomic electron density functions is given by
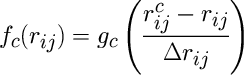
where rcij is a cutoff length and Delta_rij is the width of the cutoff region.
The indices of the parameters refer to the atom types involved. The following IMD parameters correspond to the MEAM potential parameters:
| meam_cmin | C0kij |
| meam_cmax | C1kij |
| meam_t1 | t(1)i |
| meam_t2 | t(2)i |
| meam_t3 | t(3)i |
| meam_f0 | f0i |
| meam_r0 | r0i |
| meam_beta0 | beta(0)i |
| meam_beta1 | beta(1)i |
| meam_beta2 | beta(2)i |
| meam_beta3 | beta(3)i |
| meam_deltar | Delta_rij |
| meam_rcut | rcij |
The pair potential phiij has to be given as a potential table. The corresponding IMD parameter is core_potential_file.
The embedding energy function Fi can be read in as a potential table using the IMD parameter embedding_energy_file. Alternatively, IMD computes the embedding energy analytically according to

In this case, the parameters have to be specified using the following IMD parameters:
| meam_a | Ai |
| meam_e | Eci |
| meam_rho0 | rho0i |
The atomic electron density functions rhoa(s)i(r) are computed analytically. The function rhoa(0)i(r), however, can be read in as a potential table using the IMD parameter el_density_file.
The many-body interactions of the MEAM potential are computed using neighbour tables. In constrast to the covalent potentials the IMD parameter neigh_len has the default value 12.
If more than one atom type are used, common values of the parameters meam_cmin and meam_cmax for all atom types can be chosen by specifying only one value for each parameter.
The screening function Sij can be switched off (that is, set to unity) by choosing the parameters meam_cmin and meam_cmax to be equal.